
2024年4月4日,巴西国家卫生监督局发布了一项具有里程碑意义的法规——规范性指令第290号(IN N° 290/2024)。该法规依据第741号董事会决议(RDC nº 741/2022),正式确立了医疗器械注册的优化审批程序,核心在于认可并利用等效外国监管机构的评审成果。本文件旨在深入解读该法规的细节、适用条件与操作流程,为国际医疗器械制造商提供进入巴西市场的加速策略。
一、法规总览与核心目标
1.法规名称:
葡萄牙语全称:Instrução Normativa – IN N° 290, de 4 de Abril de 2024.
中文译名:2024年4月4日第290号规范性指令。
2.上位法依据:本法令基于RDC nº 741, de 10 de agosto de 2022制定,该决议为ANVISA承认国际监管合作奠定了法律基础。
3.核心目标:通过认可已通过美国食品和药物管理局(FDA)、加拿大卫生部等严格监管体系批准的医疗器械的安全性、有效性和质量证据,简化其在巴西的注册流程。此举旨在:
-加速创新产品上市:缩短在巴西的审批时间。
-优化监管资源:使ANVISA能聚焦于更高风险或尚无国际参考的新产品。
-促进患者可及性:让巴西患者更快获得全球先进的医疗技术。
二、法规深度解读:关键概念与适用范围
- 核心机制:利用等效外国监管机构分析
法规的核心是“Aproveitamento de Análises de AREE”,即利用等效海外监管机构的分析报告。这意味着,如果您的产品已获得指定AREE的批准,您可以向ANVISA提交该机构的评审结论、关键评估报告及批准文件,作为您技术文档的核心支撑,从而避免在巴西进行完全重复的全面技术评审。 - 等效外国监管机构
AREE通常指那些拥有与ANVISA相当的科学严谨性、监管透明度和独立性的监管机构。目前被ANVISA认可的海外监管机构:
-美国食品和药物管理局(FDA)
-加拿大卫生部(Health Canada)
-日本厚生劳动省(MHLW)
-澳大利亚治疗商品管理局(TGA) - 适用范围与产品类别
该优化程序主要适用于已通过上述AREE批准,且在巴西申请注册的III类、IV类医疗器械。
通常,以下情况可能更易被接受:
-产品在原批准国与在巴西申请的预期用途、技术规格和标签基本一致。
-产品的生产场地和质量体系已通过国际认可的标准(如ISO 13485)认证,并且符合巴西良好生产规范要求。
-提交的AREE文件完整、清晰,足以支持ANVISA的评审。
三、优化注册流程
下图为您展示了利用IN N° 290/2024法规,通过优化路径完成巴西注册的全流程,以及与常规路径的关键区别:
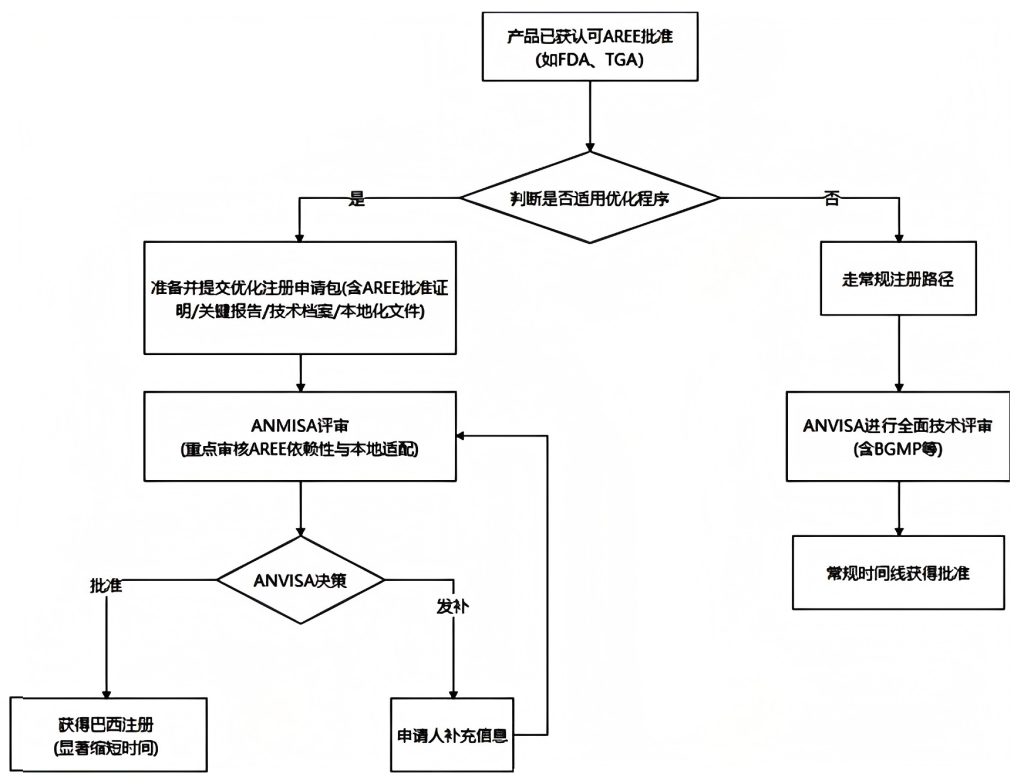
优化流程解读:
1.资格预审与规划:
(1) 确认产品已获得至少一家认可AREE的上市批准。
(2) 评估产品在巴西的申请是否与原批准的范围保持一致。
(3) 确保您的巴西注册持有人已确定并准备就绪。
2.文件准备与编译:
1) 核心AREE文件:翻译并公证AREE的批准函、决策摘要、以及任何公开或可提供的关键评估报告。
2) 技术档案摘要:准备一份指向完整技术档案的结构化摘要,并确保完整技术档案可根据要求提供。
3) 巴西特定文件:
➢符合ANVISA要求的葡萄牙语标签和使用说明书。
➢技术负责人声明。
➢证明与原批准一致性的声明信。
➢必要的本地化研究报告(如适用,包括临床或性能研究)。
3.提交与ANVISA评审:
通过ANVISA的电子提交系统(如Petições Eletrônicas)提交申请。
ANVISA的评审将聚焦于:
➢所依赖的AREE批准的有效性和充分性。
➢产品对巴西人群的适用性。
➢标签和说明书的本地合规性。
➢与巴西法规要求的其他方面(如BGMP、注册持有人协议等)的符合性。
4.互动与批准:
在评审过程中,ANVISA可能会要求解释或补充信息。及时专业的回应至关重要。
批准后,产品注册信息将在巴西官方公报上公布。
四、潜在优势、挑战与战略建议
- 优势
-显著缩短时间:预计可将常规需要12-24个月的III/IV类产品注册时间大幅缩短。
-降低注册成本:减少ANVISA全面技术评审带来的资源投入和潜在延期成本。
-提高预测性:基于明确的AREE批准,注册结果的可预测性增强。 - 挑战与注意事项
-非自动批准:AREE批准是重要依据,但ANVISA仍进行独立决策,可能提出特定要求。
-文件翻译与适配:高质量的葡萄牙语翻译和本地化适配是关键,错误可能导致评审延迟。
-法规动态性:ANVISA对AREE的认可范围和具体操作细节可能动态调整,需持续关注。
3.战略建议:
-早期评估:在产品全球开发早期,就将巴西的注册策略(尤其是利用IN N° 290的可行性)纳入考量。
-专业伙伴:与熟悉该优化程序、拥有成功经验的巴西注册持有人或本地法规顾问紧密合作。
-完整档案管理:在全球注册过程中,注意维护一份完整、清晰、易于提取的技术档案,以备巴西申请之需。
-主动沟通:在提交前,可考虑通过ANVISA的预提交渠道,就特定产品的适用性进行非正式咨询。
IN N° 290/2024的颁布是巴西监管现代化和与国际接轨的重要一步。它为代表了一个“监管桥梁”,为已获全球主要市场批准的、安全有效的医疗器械进入巴西提供了一条更高效、更可预测的路径。
对于制造商而言,成功利用此路径的关键在于:透彻理解法规要求、精心准备申请包、并与专业的本地合作伙伴协同作战。随着该法规的实施,预计将有效促进巴西医疗器械市场的创新竞争和产品可及性,为全球制造商和巴西患者带来双赢局面。


-1-300x129.png)

