
您的公司正计划拓展马来西亚业务吗?是否对琳琅满目的合规要求感到困惑?究竟哪些产品必须通过CAB认证?认证流程会不会非常漫长复杂?别担心,关于马来西亚CAB机构的种种疑问,本文将为您一一梳理清晰。
# 1. 什么是CAB机构?
在马来西亚医疗器械监管体系(Medical Device Authority, MDA)中,符合性评估机构(Conformity Assessment Body, CAB)是连接制造商与市场准入的核心枢纽。CAB认证机构是经马来西亚医疗器械管理局(MDA)注册和认可的第三方独立组织,在医疗器械监管体系中扮演着“技术守门员”的角色。其核心职责是确保医疗器械在进入马来西亚市场前符合安全、性能和质量标准。
CAB认证机构是根据马来西亚2012年医疗设备法(第737号法)由医疗设备管理局(MDA)注册和认可的组织。认证及后在医疗设备的监管框架中发挥着重要作用,通过进行各种符合性评估,确保医疗设备符合适用的标准、法规或要求。认证机构进行的符合性评估可能涵盖各种领域,包括产品安全、性能和质量管理体系。最终目标是确保投放市场上的医疗设备是安全的,并且能够按照制造商的预期进行工作,因此符合医疗设备的安全性能和性能的基本原则。
CAB通过进行各种符合性评估,确保医疗器械符合适用的标准、法规或要求,在医疗器械的监管框架中发挥着重要作用。
# 2. 什么产品需要进行符合性评估?
根据马来西亚《2012年医疗器械法》及相关法规,Class A产品通常不需要经过CAB评估,可直接向马来西亚医疗器械管理局(MDA)提交申请,即通过MeDC@St 2.0+ 系统提交注册申请。
Class B、C、D按要求在注册前必须进行符合性评估(CAB),如果Class B、C、D如果已获得马来西亚认可国家(如美国FDA、欧盟公告机构、日本、泰国、新加坡、澳大利亚、英国等)的批准,CAB通常采用验证方式(Verification)进行简化评估;否则需进行全面符合性评估。
认可的监管机构或公告机构清单:
| 欧盟/欧盟公告机构(EU NB) | 对于 B 类,根据以下法规签发的 EC 证书:・指令 93/42/EEC 附录 II 第 3 节或附录 V(适用于 IIa 类医疗器械设备)・医疗器械法规(MDR)附录 IX 第一章和第三章,或 MDR 附录 XI A 部分(适用于 IIa 类产品)・指令 98/79/EC 附录 IV 或附录 V 及附录 VII(适用于 B 类和自我检测类体外诊断医疗器械)・体外诊断医疗器械法规(IVDR)附录 IX 第一章和第三章(适用于 B 类体外诊断医疗器械) 对于 C 和 D 类,根据以下法规签发的 EC 证书:・指令 93/42/EEC 附录 II 第 3 节或附录 III 及附录 V(适用于 IIb 类)・医疗器械法规(MDR)附录 IX 第一章和第三章,包括技术文件评估(适用于植入式医疗器械),或 MDR 附录 X 及附录 XI A 部分(适用于 IIb 类)・指令 93/42/EEC 附录 II 第 3 和第 4 节(适用于 III 类)・医疗器械法规(MDR)附录 IX 第一章和第三章,包括技术文件评估(适用于 III 类)・指令 90/385/EEC 附录 II 第 3 和第 4 节适用于有源可植入医疗器械,(注:指令 90/385/EEC 已纳入 MDR,有源可植入医疗器械在 MDR 下为 III 类)・指令 98/79/EC 附录 IV(包括第 4 和第 6 节,适用于 A 类体外诊断医疗器械)・IVDR 附录 IX 第一章和第三章,包括技术文件评估(适用于 D 类体外诊断医疗器械)・指令 98/79/EC 附录 IV 或附录 V 及附录 VII(适用于 B 类和自我检测类体外诊断医疗器械)・体外诊断医疗器械法规(IVDR)附录 IX 第一章和第三章,包括伴随诊断、自测及近患者检测器械的技术文件评估,或 IVDR 附录 X 及附录 XI(第 5 节除外,适用于 C 类体外诊断医疗器械) |
| 日本厚生劳动省(MHLW) | 日本注册认证机构的上市前认证(Ninsho)厚生劳动省的上市前批准(Shonin) |
| 澳大利亚/治疗用品管理局(TGA) | ARTG注册证书 |
| 加拿大卫生部(HC) | 加拿大卫生部许可证 |
| 美国食品药品监督管理局(US FDA) | 510K许可上市前的批准(PMA) |
| 英国药品和医疗产品监管局(MHRA) | 英国而言:英国合格评定(UKCA)根据上述欧盟NB中列出的公认批准签发的EC证书对于北爱尔兰:根据上述欧盟NB中列出的公认批准签发的EC证书英国北爱尔兰(UKNI)和根据上述欧盟 NB 中列出的公认批准签发的 EC 证书 |
| 新加坡卫生科学管理局(HSA) | 在新加坡医疗器械注册处(SMDR)注册 |
| 泰国食品药品监督管理局(FDA) | II-III类:通知类医疗器械证书IV类:许可类医疗器械证书 |
# 3. 符合性评估的路径有哪些?
符合性评估的路径主要分为全流程评估路径、验证评估(初次认证评估路径、再认证评估路径)。
下面介绍一下路径的具体要求:
| 合规评定路径 | 医疗器械认证资格条件 |
| 全流程 | 该医疗器械设备未从任何公认的监管机构或通知机构获得批准。 |
| 验证(初始认证评估) | 该医疗器械;1. 已获得至少一个认可的监管机构或认证机构的批准;2. 其设计和预期用途与已获得认可监管机构或认证机构批准的医疗器械相同。3. 在过去的一年里,当按照制造商的预期方式使用该医疗设备时,未发现任何全球性的安全问题,定义如下:1)无死亡报告;2)未发现任何人健康状况出现严重恶化的情况。并且3)在提交时未采取任何开放式的安全纠正措施(包括召回措施);并且注意:所有事故报告以及纠正和预防措施均已关闭。4. 尚未被任何公认的监管机构或认证机构拒绝或撤销。 |
| 验证(再认证评估) | 该医疗器械;· 已通过完全一致性评估其合规性,评估或验证途径已向MDA注册和在CAB进行再认证评估前,已拥有有效的注册证书;· 设计、规格、特性或注册相关信息未发生任何变更,除非已通知MDA; · 未遭MDA拒绝或撤销注册 |
若从认可监管机构或通知机构获得的批准被撤销或失效,医疗器械应进行全面的符合性评估。
需要注意的是验证途径不适用于根据以下方案/计划获得特殊授权进入市场的医疗器械,包括但不限于下表中列出的情况:
| 国家/监管机构 | 方案/程序 |
| 美国/食品药品监督管理局(FDA) | 紧急使用授权(EUA)扩展使用(紧急/同情性使用)人道主义设备豁免(HDE) |
| 欧盟/国家主管当局(EU) | 特殊使用授权 |
| 英国/药品和医疗产品监管局(MHRA) | 特殊使用授权 |
| 加拿大/卫生部 | 特殊获取计划(SAP)*临时命令(IO) |
| 日本/药品医疗器械局(PMDA) | 同情使用制度*紧急监管路径 |
| 澳大利亚/治疗用品管理局(TGA) | 特殊获取计划(SAS)授权处方者计划、*紧急豁免 |
| 新加坡/卫生科学管理局(HSA) | 特殊获取途径 |
| 泰国/食品药品监督管理局(TFDA) | 《医疗器械法》第27条规定的非商业用途医疗器械生产或进口豁免 |
注:
1)这些方案/计划为未经批准的医疗器械提供了临时的使用授权,使其得以进入市场。紧急用途市场,旨在确保患者在面临危急或紧急情况时能够获得必要的医疗设备。
2)*在新冠疫情期间,获得特别批准/许可以进入市场。
# 4. 进行符合性评估的流程是什么?
符合性评估流程图:
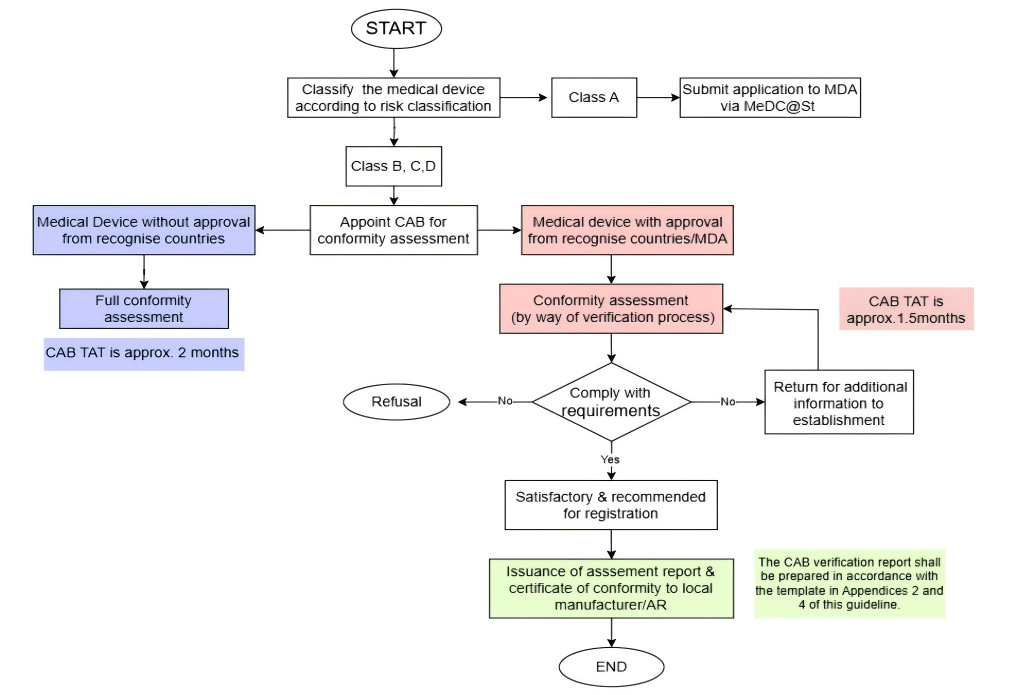
# 5. 进行符合性评估所需的完整评估周期是多久?
CAB进行符合性评估所需的完整评估时间(TAT)时间表如下表所述。此时间表不包括在提交过程中任何响应的准备时间(仅供参考)。
初始及重新认证流程的周转时间(TAT)
| 类别 | 流程阶段 | 首次/再认证 |
| 初始CAB-建立委托 | 建立业务关系 | 3个工作日 |
| 初始申请提交 | 3个工作日 | |
| 合同审查 | 3个工作日(2周内完成) | |
| 预审准备 | 预审核文件准备 | 机构响应 |
| 文件提交 | 机构响应 | |
| 安排 | 3个工作日 | |
| 提交文件以供审核 | 初步文件审查 | 每个医疗设备4个小时 |
| 认证决定 | 最终审查&认证决定 | 报告最终提交后两周内完成审核并作出决定 |
| 认证决定通知 | 5个工作日 | |
| 证书颁发后的操作步骤 | 认证费用支付 | 按照双方约定的时间 |
| 证书发放 | 费用支付后的2个工作日 | |
| 整体时间(月) | 预计1.5月 | |
如果有更多关于注册的问题,欢迎联系我们!



于2025年12月17日发布的《医疗器械注册手册》-300x191.png)
