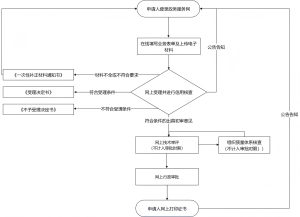
一、档案格式与CSDT要求
肯尼亚PPB要求所有医疗器械注册档案采用通用提交档案模板(Common Submission Dossier Template, CSDT)格式,该模板融合了IMDRF的STED框架。档案必须包含:
· 管理信息
· 制造商信息
· 设计验证与确认文件
· 风险分析
· 临床评估报告
· 上市后监督计划
· 生物安全性数据(如适用)
· 灭菌验证报告(如适用)
未按CSDT格式提交的申请可能被退回或延迟评审。
二、本地授权代表(LAR)的核心责任
境外制造商必须指定一个在肯尼亚的本地授权代表,其法律责任贯穿产品全周期:
· 作为与PPB的主要联络点
· 保存技术文件供监管机构审查
· 负责不良事件与事故报告
· 确保供应链合规
· 执行现场安全纠正措施
· 协助提交变更通知
同一产品不得授权多个LAR,否则注册无效。
三、分类规则与多重类别处理
医疗器械分类由制造商依据其预期用途和风险特征确定。若设备符合多个分类规则,则按最高风险类别执行。
例如,既是侵入式又具药物释放功能的设备,通常归类为D类。分类一旦确定,需在申报材料中提供详细理由。
四、加急与即时注册的准入条件
肯尼亚为B类设备提供即时注册(IBR)路径,条件严格:
· 已获至少三个参考监管机构批准(如FDA、CE、TGA)
· 在至少两个市场上市满4年
· 过去3年无死亡、严重健康恶化或未结案召回事件
· 无因质量/安全问题被任何监管机构拒绝或撤销的记录
符合条件者可在线提交后1小时内完成注册,极大缩短上市时间。
五、变更管理的分类与报批
任何影响设备安全性、质量或功效的变更均需通知PPB:
· 技术变更(C/D类):需事先批准
· 评审变更(A/B类):如用途扩展、新增型号、警告内容修订等,也需事先批准
· 通知变更:如标签文字调整,提交后即可实施(安全相关除外)
变更分类错误可能导致注册暂停或撤销。
六、上市后监督与年度保留
注册后企业必须建立并执行上市后监督(PMS)计划,收集安全与性能数据。此外,每年需在到期前60天内支付年度保留费,否则注册将被暂停或取消。
七、标签与使用说明的合规要点
· 标签须包含制造商名称、地址、批号、有效期(如适用)、预期用途等
· 使用说明需清晰描述操作步骤、警告、禁忌症
· 若设备含软件,需提供软件生命周期文档和验证报告
如果您在过程中需要专业的支持与服务,欢迎随时联系我们。

于2025年12月17日发布的《医疗器械注册手册》-300x191.png)