于2025年12月17日发布的《医疗器械注册手册》.png)
本文解读的是巴西国家卫生监督局(ANVISA)于2025年12月17日发布的《医疗器械注册手册》(MANUAL PARA REGISTRO DE MATERIAIS DE USO EM SAUDE NA ANVISA)。该手册由ANVISA的医疗器械技术管理处(GEMAT)编写,旨在整合最新法规修订,为医疗器械制造商和进口商提供清晰的注册流程指导。手册基于多项关键法规更新,包括RDC n° 751/2022、RDC n° 556/2021和RDC n° 848/2024等,重点修订了风险分类、注册程序和安全要求等内容。以下将详细解析核心修订内容、发布日期与实施日期,并为制造商和进口商总结申请材料准备的关键指导方针。
一、核心修订内容详解
本手册的核心修订主要基于以下ANVISA法规,这些法规对医疗器械的注册框架进行了重大调整:
1.风险分类系统的更新(依据RDC n° 751/2022)
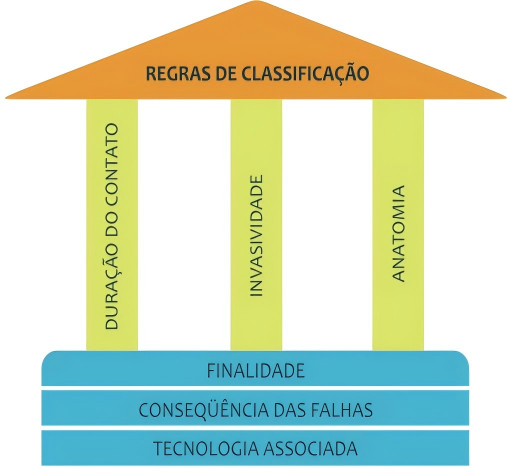
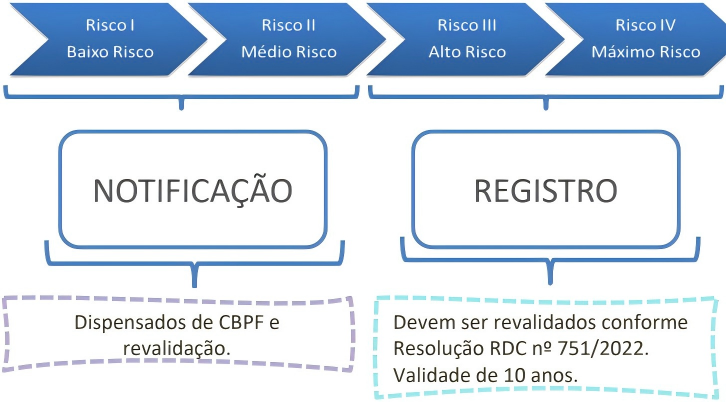
- 修订要点:RDC n° 751/2022于2022年9月15日发布,彻底改革了医疗器械的风险分类体系。医疗器械根据固有风险分为I类(低风险)、II类(中风险)、III类(高风险)和IV类(最高风险)。I类和II类产品适用通知制度(notificação),而III类和IV类必须进行注册(registro)。
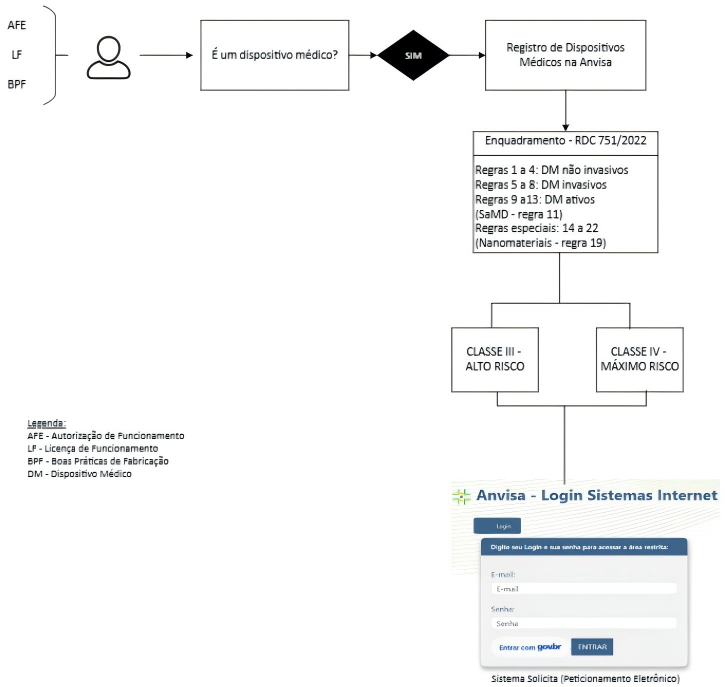
- 具体规则:分类基于22条规则,考虑因素包括使用时间(短暂使用、短期使用、长期使用)、侵入性(非侵入性、侵入性、植入性)以及设备类型(主动设备、特殊设备等)。例如,规则8规定,接触中枢循环或神经系统的设备自动归为IV类。
- 影响:这一修订使分类更精细化,要求制造商在申请前必须准确评估产品风险,否则可能导致申请被拒。
2.注册程序的优化与电子化
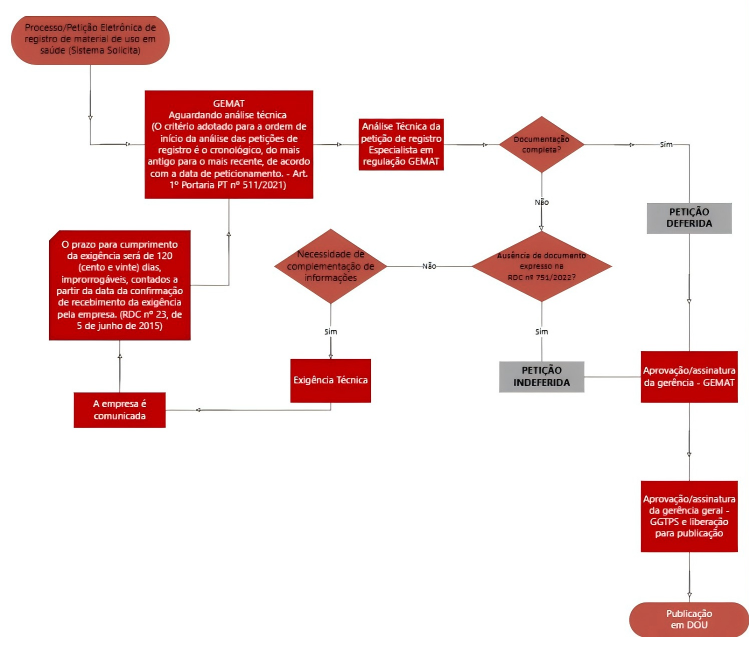
- 修订要点:手册强调电子申请为唯一渠道,通过ANVISA的Solicita或Peticionamento系统提交。新流程引入了“优化分析程序”(procedimento otimizado),允许利用等效外国监管机构(AREE)的评估结果,加速III类和IV类设备的注册(依据IN n°290/2024)。
- 关键变化:注册有效期统一延长:根据自2022年9月起生效的RDC n°751/2022,医疗器械注册证书的有效期已统一延长至10年(原为5年)。此次手册是对这一既定规则的再次明确和强调。再验证申请必须在有效期届满前6-12个月内提交。注册号格式标准化为11位数字(前7位为AFE号,后4位为序列号),增强了产品的可追溯性。
3.安全与性能要求的强化(依据RDC n° 848/2024)
- 修订要点:RDC n° 848/2024于2024年3月6日发布,明确了医疗器械必须满足基本安全和性能要求(requisitos essenciais de segurança e desempenho)。制造商需提交技术档案(Dossiê Técnico),包括风险管理报告、临床评估数据和生物相容性测试结果。
- 新增要求:对于创新型设备,必须提供特定临床研究数据。此外,标签和说明书必须包含唯一设备标识(UDI)信息,以增强可追溯性。
4.分组和家族注册的明确化(依据RDC n° 556/2021)
- 修订要点:该法规允许将相似设备分组为家族、系统或套装进行注册,减少重复申请。但要求组内设备必须具有相同设计和用途。
- 注意事项:如果初始注册时未选择家族模式,后续无法更改,必须重新申请。
二、发布日期与实施日期
手册发布日期:本手册由ANVISA于2025年在巴西利亚发布,作为最新指导文件,替代以往版本。
关键法规的发布日期与实施日期:
- RDC n° 751/2022:发布日期为2022年9月15日,自公布之日起生效,手册基于其内容更新。
- RDC n° 556/2021:发布日期为2021年8月30日,立即实施。
- RDC n° 848/2024:发布日期为2024年3月6日,生效日期同公布日。
- IN n° 290/2024:发布日期为2024年4月4日,用于规范AREE优化程序。
实施注意事项:手册本身无强制力,但引用的法规具有法律效力。企业需确保申请流程符合最新法规,否则可能面临申请被拒或处罚。
三、医疗器械制造商和进口商申请材料准备指导方针
为正确准备注册申请材料,手册为制造商和进口商提供了以下明确指导方针,涵盖从公司注册到文档提交的全流程:
1.公司合规前置要求
- 企业运营授权(AFE):制造商或进口商必须先获得ANVISA颁发的AFE,证明企业有资质从事医疗器械相关活动。申请需通过电子系统提交,依据RDC n°16/2014。
- 地方卫生许可(LF):企业还需从地方卫生局(VISA)获取运营许可证,确保设施符合标准。
- 良好生产规范证书(CBPF):对于III类和IV类设备,必须持有ANVISA颁发的CBPF或至少提交申请协议。依据RDC n° 687/2022,CBPF是注册批准的前提条件。
2.申请材料的核心文档清单
申请材料必须通过电子系统提交,以下为关键文档要求(基于技术档案结构):
- 申请表(Formulário de Petição):需完整填写设备名称、型号、风险分类等信息。表格不可修改,所有字段必须准确无误。
- 技术档案(Dossiê Técnico):分为6章,需包括:
第1章:管理和技术信息(如公司数据和产品标识)。
第2章:产品详细描述、全球商业历史和使用目的。
第3章:安全和性能数据,如风险管理报告、物理化学特性、生物相容性测试(依据ISO 10993标准)、灭菌验证等。
第4章:临床证据摘要,需符合IMDRF指南,证明设备安全有效。
第5章:标签和说明书模型,必须用葡萄牙语编写,包含UDI信息。
第6章:生产流程流程图,展示从原材料到成品的所有步骤。
附加文档:
•对于进口设备:需提供制造商授权书(授权巴西代表)和自由贸易证书(CLC),均需公证或海牙认证,并在2年内有效。
•特定设备要求:如植入式设备需提供UDI标签;无菌设备需有灭菌方法说明。
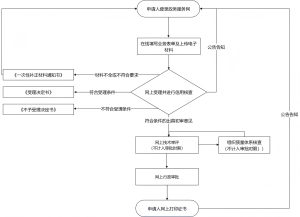
3.申请流程分步指导
步骤1:公司注册和规模确认(通过ANVISA系统完成cadastramento)。
步骤2:选择申请主题(Código de Assunto),如“Solicitar Registro de Dispositivos Médicos”,并核对文档清单。
步骤3:电子提交申请并支付监管税(TFVS),税率基于公司规模和产品类型。
步骤4:协议提交后,在120天内回应任何技术质询(exigência técnica),否则申请可能被拒。
步骤5:批准后,注册公告发布于联邦官方公报(DOU),设备方可上市。
4.常见错误与避免建议
文档不全:如缺少CBPF或临床报告,会导致直接拒绝。建议使用ANVISA网站上的核对清单(checklist)。
分类错误:错误风险分类是常见拒绝原因。企业可提前通过ANVISA咨询渠道确认分类。
标签不符:说明书必须与技术档案一致,避免使用误导性信息。
四、结论
2025版ANVISA手册整合了关键法规更新,提供了清晰的战略蓝图。其核心价值在于:
1.提升服务标准与效率
手册的标准化要求(如电子化流程、AREE快速通道)使我们能为客户规划更高效的注册路径,显著提升项目成功率与时效性。
2.深化专业服务价值
详细的技术档案与临床证据要求,推动我们的服务从文件代理深化为全方位的合规战略伙伴,确保客户产品的安全性与有效性。
3.保障客户市场准入
精通手册是我们为客户降低合规风险、加速市场准入的核心。我们将持续追踪法规动态,确保客户项目的长期合规活力。
总之,本手册是我们将复杂法规转化为客户明确市场竞争优势的关键工具,助力其在巴西市场稳健发展。
如果您需要专业的支持与服务,欢迎随时联系我们。